
Medical Devices
A certificate of compliance with ISO-13485 (Medical Devices – Quality Management Systems – Requirements... View more
Organizer:
- Organized by
-
-
21 CFR 812 – What Medical Device Companies Need to Know
-
21 CFR 812 – What Medical Device Companies Need to Know
What is 21 cfr 812 and how should it be used?
A guidance document represents the FDA’s current thinking on a topic. Per FDA’s website, guidance documents “do not create or confer any rights for or on any person and do not operate to bind FDA or the public. You can use an alternative approach if the approach satisfies the requirements of the applicable statutes and regulations.” Guidance documents usually discuss FDA’s interpretation of their policy on a regulatory issue.

One way to think about guidance documents are as suggested opinions of best practices from the FDA. Guidance documents work hand-in-hand with the applicable regulations (21 CFR 812, 312, 50, 54, 56) to deliver recommendations of leading best practices to produce clinical research in a manner that protects patient’s rights, safety, and welfare while delivering clinical data with integrity. The phrase “can use an alternate approach” may seem confusing, but this allows for interpretation of the guidance document with respect to the conduct of clinical studies, as long as the requirements of the regulations are satisfied.
The FDA recently created a database for all guidance documents with the goal of creating a convenient way to search all FDA guidance documents from a single location. This database can be accessed through their publicly available website. It allows for searching based on product, date issued, FDA organization, document type, subject, draft or final status, open for comment, and comment closing date for draft guidances. Also, a Regulatory Affairs Professional Society (RAPS) article stated that the database is configured to eventually include information from FDA’s operation manuals, compliance policy guides, import alerts, and more. Importantly, it was recently noted that the database includes many but not all guidance documents yet. Stay tuned for updates on this key resource.
21 CFR 812 & A PRINCIPAL INVESTIGATOR (PI)’S OVERSIGHT
According to the regulations for clinical trials, the PI is personally responsible for conducting 21 CFR 812 and supervising the conduct of human subjects research by “protecting the rights, safety, and welfare of subjects under the investigator’s care”. The PI also has to ensure that all the research he is responsible for is conducted in an ethical manner and in accordance with all federal, state, and local laws and regulations, institutional policies, and the requirements of the IRB. So what exactly is “oversight”? Oversight is defined in the Merriam-Webster’s online dictionary as “management by overseeing the performance or operation of a person or group; watchful care, superintendence, general supervision”. As you can imagine, this can be quite an undertaking.
In order to provide oversight and to ensure that the rights, safety, and welfare of research subjects is protected the PI must ensure that (among other things):
He/she or another qualified individual provides study subjects with reasonable medical care for any adverse events, including clinically significant laboratory values, related to the research
He/she or another qualified individual is available to study subject to answer questions or provide care during the conduct of the research
All research staff adhere to the research plan (i.e., inclusion/exclusion criteria, safety assessments, safety monitoring and reporting of unanticipated problems)

There is protection of privacy and confidentiality of identifiable data
There is minimization of risks to the subjects
An investigator also is responsible for ensuring that informed consent is obtained in accordance with the regulations
It is the PI’s responsibility to not begin any research study without adequate resources to protect the subjects who are participating. The PI should have a plan for supervision of all tasks throughout the investigation. The FDA offers Guidance on Investigator Responsibilities for suggestions on how to take on all this responsibility. With so many regulations from various entities to abide by, it becomes clear why oversight can sometimes become “an oversight”.
Rule and Guidance on Acceptance of Clinical Data to Support Medical Device Applications and Submissions
The new rule seeks to provide a consistent approach to acceptance of clinical data, regardless of whether the data were collected inside or outside the United States (OUS). To this end, however, the amendments mostly affect the requirements for clinical investigations conducted OUS. As noted in FDA’s guidance, these amendments appear to be, in part, the result of sponsors’ increased reliance on data from clinical investigations conducted OUS.
For Investigational Device Exemption (IDE) applications or premarket submissions supported by clinical data from investigations conducted OUS, the final rule adds a new provision to the IDE regulations (21 C.F.R. § 812.28) to ensure that the investigations comply with good clinical practices (GCP). This new provision states that FDA will accept data from a clinical investigation conducted OUS if the investigation is “well-designed and well-conducted” and the following information is provided:
- A statement that the investigation was conducted in accordance with GCP;
- The names of the investigators and the names and addresses of the research facilities and sites where records relating to the investigation are maintained;
- The investigator’s qualifications;
- A description of the research facility;
- A detailed summary of the protocol and results;
- Either a statement that the device used in the investigation is identical to the device that is the subject of the submission, or a detailed description of the device used in the investigation and a comparison to the device that is the subject of the submission;
- If the investigation is designed to support the safety and effectiveness of a device, a discussion demonstrating that the data constitute valid scientific evidence within the meaning of 21 C.F.R. § 860.7;
- The name and address of the Independent Ethics Committee (IEC) that reviewed the investigation;
- A summary of the IEC’s decision to approve or modify and approve the investigation;
- A description of how informed consent was obtained;
- A description of any incentives provided to the subjects to participate;
- A description of how the sponsor monitored the investigation; and
- A description of how investigators were trained to comply with GCP and to conduct the investigation in accordance with the protocol.
How to Get FDA Approval for Medical Devices
To get FDA approval for your medical device, you’ll need to go through the following five steps.
1. Know Your Device’s Classification
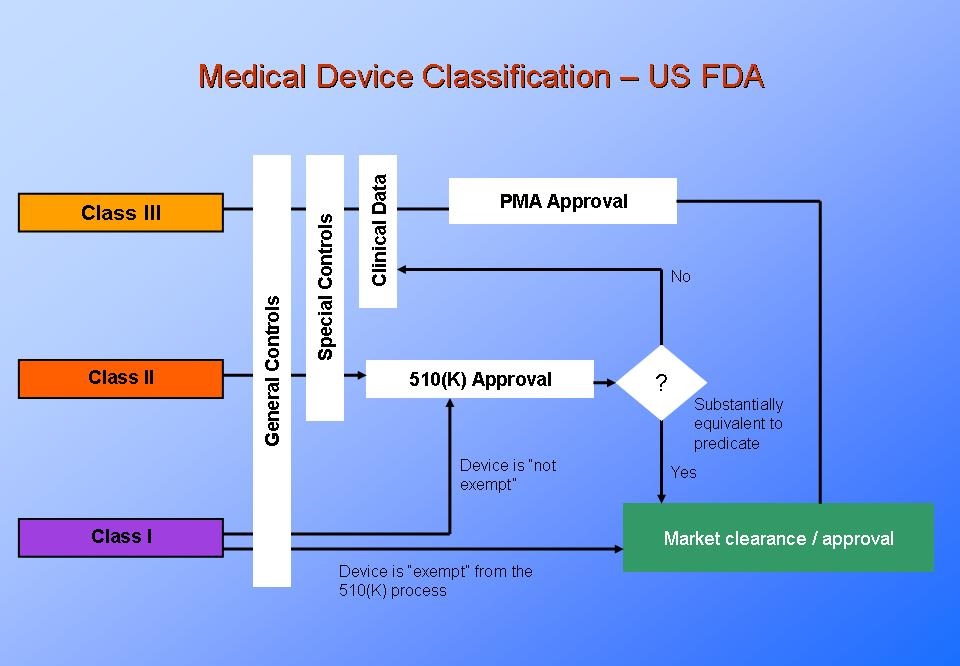
Medical devices fall into three classes:
- Class I
- Class II
- Class III

You should know your device’s classification before the development process begins. The class of device will correlate to how you’ll need to manage requirements and testing.
The class of device will also determine which regulations you’ll need to comply with. Class I medical devices are the lowest risk. So, they’re generally not subject to a pre-market review. However, Class II and Class III devices are subject to pre-market reviews.
2. Develop a Prototype
The next step is to develop a prototype. This prototype won’t be ready for human use. Instead, its main use is for testing in laboratory environments — and sufficiently reducing risk before it is used by humans. It may then go into clinical trials.
3. Submit Your Applications (If Applicable)
To get FDA clearance to market and distribute your class II or class III device, you’ll need to submit a pre-market notification or application.
As part of this, you’ll need FDA verification and validation of your device.
FDA Verification
FDA verification is important for making sure the requirements you set are fulfilled. You should be able to answer the question…
Are you designing the device right?
You can answer yes if:
- Requirements are implemented.
- Requirements are verified.
- Requirements of standards are met.
FDA Validation
FDA validation is important for making sure the device you deliver fulfills the needs of the market. You should be able to answer the question…
Are you designing the right device?
You can answer yes if your device meets the needs of:
- Device users.
- Stakeholders within your company.
- Regulatory stakeholders.
4. Wait for FDA Review and Approval
The next step is to wait for the FDA to review and approve your device.
5. Maintain FDA Compliance
You’re not in the clear once you’ve received FDA approval. You’ll need to ensure your device stays in compliance for its lifespan.


Sorry, there were no replies found.