
Medical Devices
A certificate of compliance with ISO-13485 (Medical Devices – Quality Management Systems – Requirements... View more
Organizer:
- Organized by
-
-
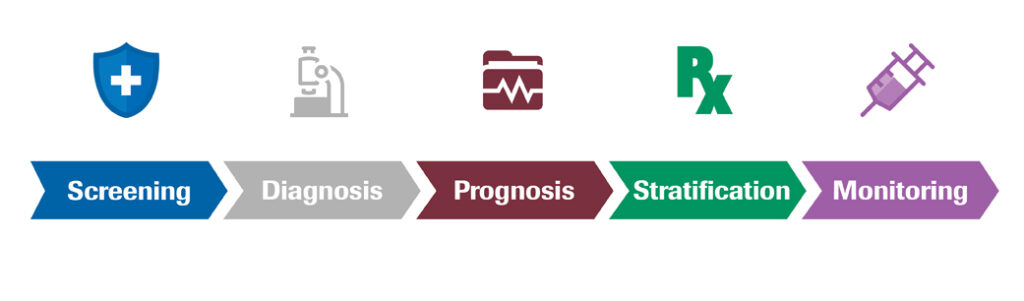
IVD (In Vitro Diagnostic) Products: Overview, Definitions and Regulation
-
IVD (In Vitro Diagnostic) Products: Overview, Definitions and Regulation
A medical device is an in vitro diagnostic medical device (IVD) if it is a reagent, calibrator, control material, kit, specimen receptacle, software, instrument, apparatus, equipment or system, whether used alone or in combination with other diagnostic goods for in vitro use. It must be intended by the manufacturer to be used in vitro for the examination of specimens derived from the human body, solely or principally for the purpose of giving information about a physiological or pathological state, a congenital abnormality or to determine the safety and compatibility with a potential recipient, or to monitor therapeutic measures.

What is an In Vitro Diagnostic product (IVD)?
Definition: In vitro diagnostic products are those reagents, instruments, and systems intended for use in diagnosis of disease or other conditions, including a determination of the state of health, in order to cure, mitigate, treat, or prevent disease or its sequelae. Such products are intended for use in the collection, preparation, and examination of specimens taken from the human body.
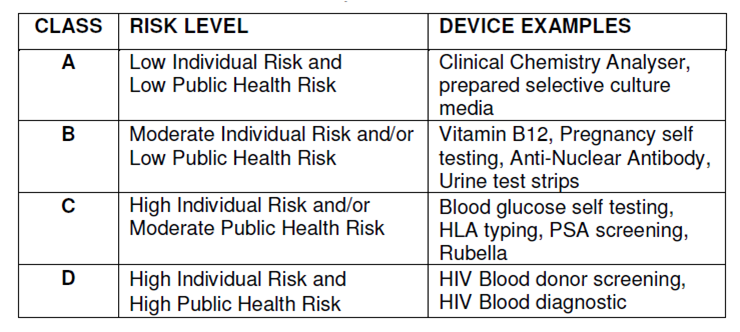
How are IVDs classified?
The FDA classifies medical devices, including IVD products, into Class I, II, or III according to the level of regulatory control that is necessary to reasonably assure safety and effectiveness. The classification of an IVD (or other medical device) determines the appropriate premarket process.

What is a General Purpose Reagent?
A general purpose reagent (GPR) is “a chemical reagent that has general laboratory application, is used to collect, prepare, and examine specimens from the human body for diagnostic purposes, and is not labeled or otherwise intended for a specific diagnostic application [General purpose reagents] do not include laboratory machinery, automated or powered systems.”
Getting your IVD medical device to market
The BSI In Vitro CE marking and Quality Management Services are designed to assist you in getting your safe and effective device to global markets efficiently. Our dedicated in-house IVD team has both industry and regulatory experience, resulting in a comprehensive understanding of your needs and challenges. We provide open communication, speed-to-market programs, and balance independence with partnership to ensure reviews stand up to scrutiny.
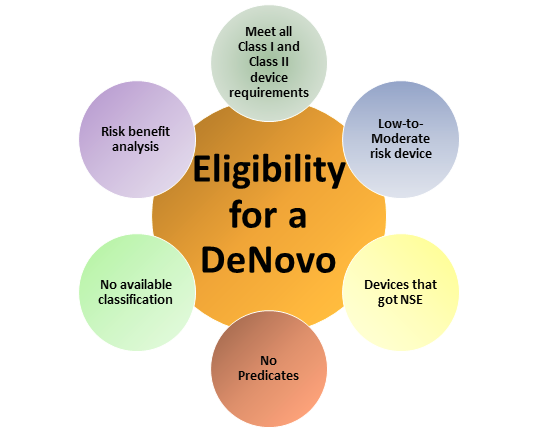
What is a De Novo Classification for IVD Devices?
The De Novo process provides a pathway to classify novel medical devices for which general controls alone, or general and special controls, provide reasonable assurance of safety and effectiveness for the intended use, but for which there is no legally marketed predicate device. De Novo classification is a risk-based classification process.

Devices that are classified into class I or class II through a De Novo Classification Request (De Novo request) may be marketed and used as predicates for future premarket notification [510(k)] submissions, if necessary.
What are the Requirements for IVD Labeling?
In Vitro Diagnostic Products have additional labeling requirements under 21 CFR 809, Subpart B, In Vitro Diagnostic Products for Human Use. Before a manufacturer obtains marketing authorization for an IVD product, they must label the product in accordance with labeling regulations.
How does the FDA ensure compliance with the Federal Food, Drug, and Cosmetic Act for IVD devices?
The FDA works with manufacturers throughout the total product life cycle to ensure compliance with the FD&C Act in the least burdensome manner for medical devices. This approach enables manufacturers to receive assistance and feedback in a timely manner and may reduce the need for compliance actions.
Sorry, there were no replies found.